Laboratory of AI Physical Sciences
A cutting-edge and comprehensive open platform that combines extensive electronic structure databases with advanced AI large model technologies, dedicated to intelligent materials design and electronic structure prediction.
Try Now
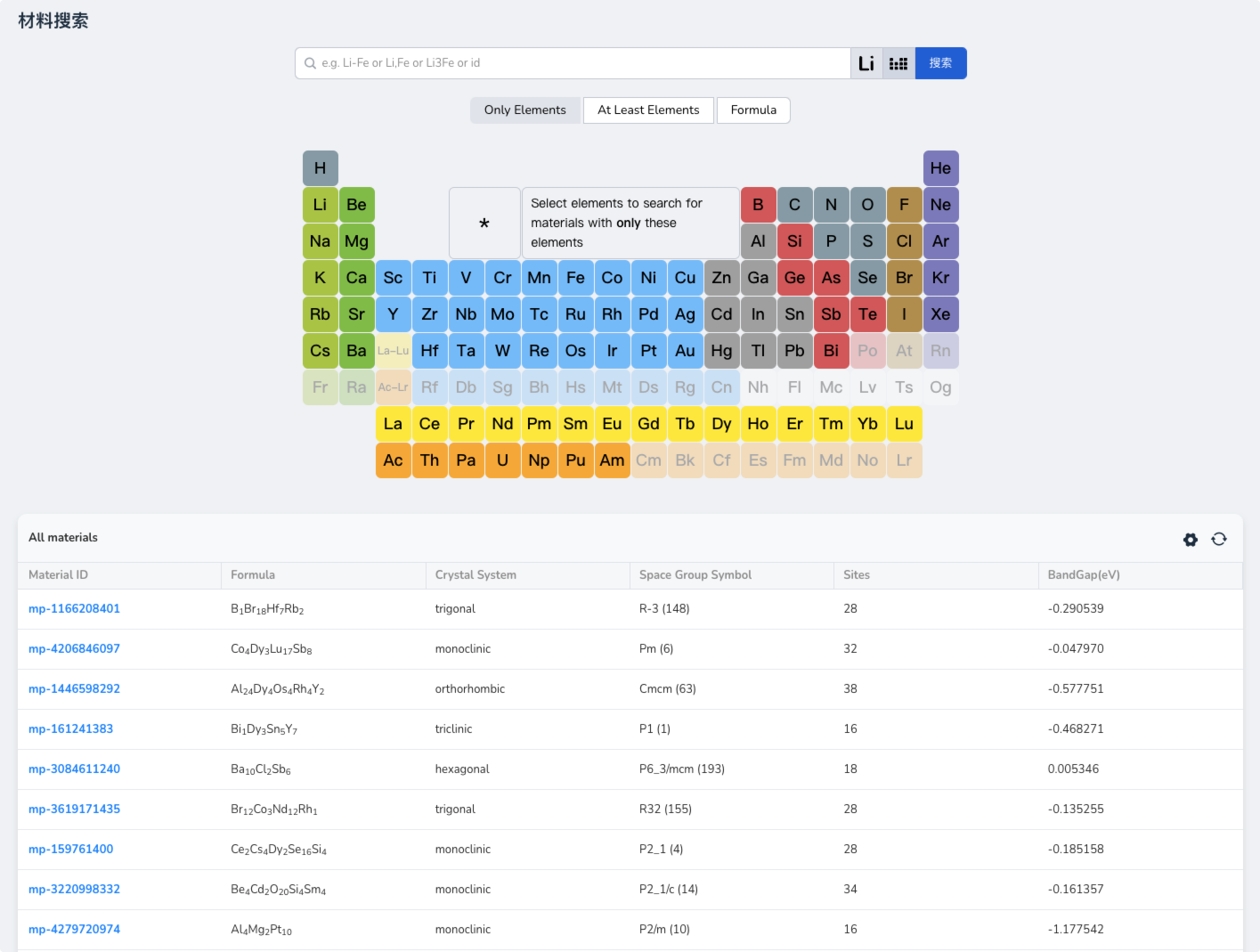
Rapid Search Based on Element Composition
The platform includes electronic structure data of over 200,000 stable crystals and provides an intuitive search function that allows users to quickly find detailed information about materials based on element composition, including 3D crystal structures, chemical formulas, and machine learning-predicted band structures and charge density distributions.
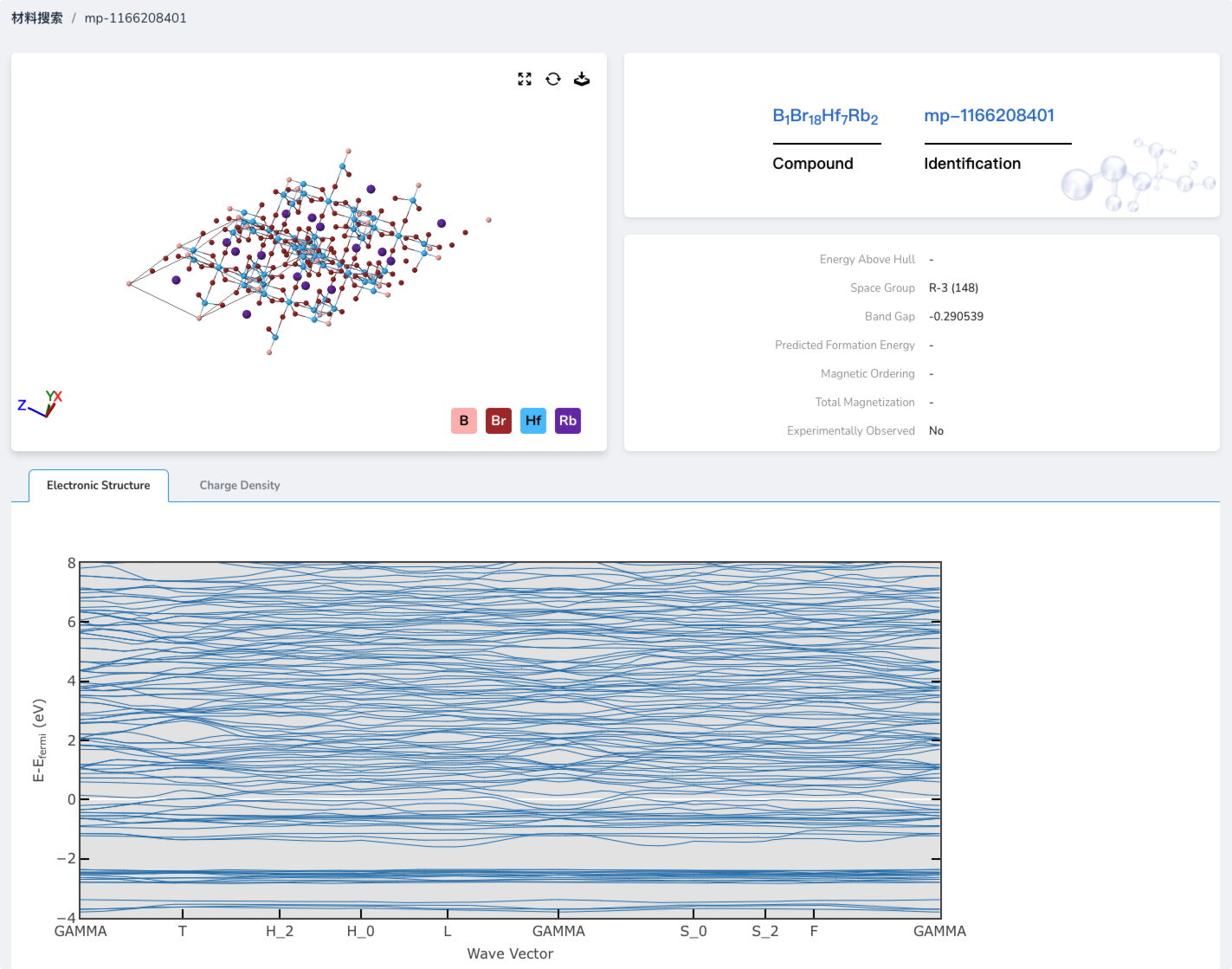
Efficient Prediction with Large Electronic Structure Models
At the core of the platform is a set of universal large electronic structure models that can efficiently predict the electronic structures of complex multi-element lattices across the periodic table. The platform also provides customized models optimized for specific systems to ensure high-accuracy results in specialized domains.
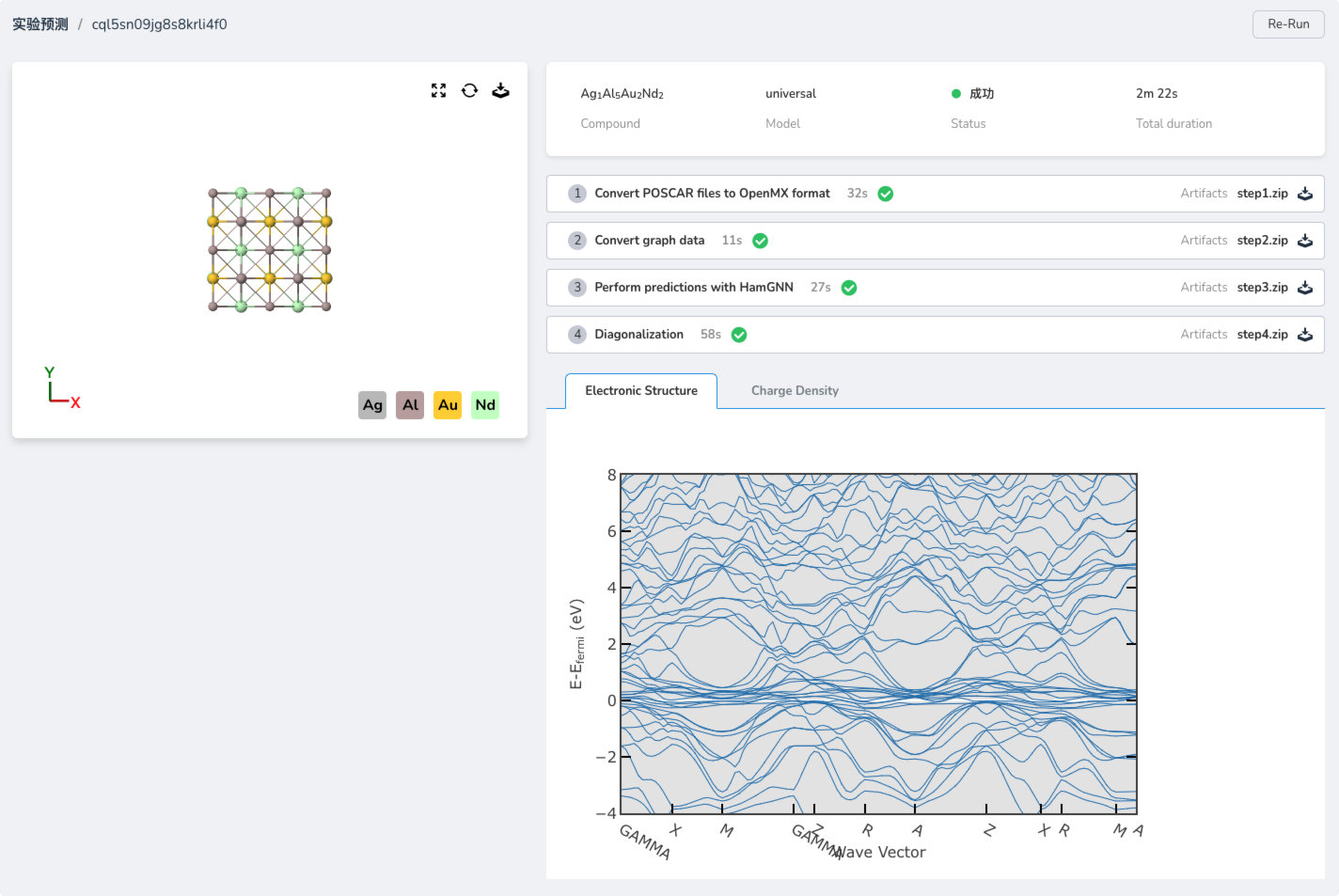
Enabling Fully Automated Experimental Workflows
Through an integrated experimental environment, users can easily upload crystal structures, configure computing resources, and automate the entire workflow—from data preprocessing to result analysis—greatly accelerating research in materials science.

This innovative platform represents a new paradigm in electronic structure calculations, with the potential to drive major breakthroughs in superconductivity, integrated circuits, data storage, and solar cells. It provides a powerful toolbox for researchers worldwide, accelerating high-throughput materials design and discovery.
AI Materials Science Lab — The Intelligent Accelerator for Materials Research
Meet the Team









Research Papers
[1] ETGNN: Zhong, Y., Yu, H., Gong, X. & Xiang, H. A General Tensor Prediction Framework Based on Graph Neural Networks, J. Phys. Chem. Lett. 14, 6339−6348 (2023).
DOI: 10.1021/acs.jpclett.3c01200[2] HamGNN: Zhong, Y., Yu, H., Su, M., Gong, X. & Xiang, H. Transferable equivariant graph neural networks for the Hamiltonians of molecules and solids, npj Comput. Mater. 9, 182 (2023).
DOI: 10.1038/s41524-023-01130-4[3] Universal Hamiltonian model: Y. Zhong, H. Yu, J. Yang, X. Guo, H. Xiang and X. Gong. Universal Machine Learning Kohn-Sham Hamiltonian for Materials, Chin. Phys. Lett. 41, 077103 (2024).
DOI: 10.1088/0256-307X/41/7/077103[4] HamEPC: Y. Zhong, S. Liu, B. Zhang, Z. Tao, Y. Sun, W. Chu, X. G. Gong, J. H. Yang, and H. Xiang. Accelerating the calculation of electron-phonon coupling strength with machine learning, Nat. Comput. Sci. 4, 615–625 (2024).
DOI: 10.1038/s43588-024-00668-7[5] Magnetic Hamiltonian model: Y. Zhong, B. Zhang, H. Yu, X. Gong, and H. Xiang, arXiv:2306.01558 (2023).
arXiv:2306.01558[6] SpinGNN++: H. Yu, B. Liu, Y. Zhong, L. Hong, J. Ji, C. Xu, X. Gong, and H. Xiang. Physics-informed time-reversal equivariant neural network potential for magnetic materials, Phys. Rev. B 110, 104427 (2024).
DOI: 10.1103/PhysRevB.110.104427[7] SpinGNN: H. Yu, Y. Zhong, L. Hong, C. Xu, W. Ren, X. Gong, and H. Xiang. Spin-dependent graph neural network potential for magnetic materials, Phys. Rev. B 109, 144426 (2024).
DOI: 10.1103/PhysRevB.109.144426[8] Spin NN Hamiltonian: H. Yu, C. Xu, X. Li, F. Lou, L. Bellaiche, Z. Hu, X. Gong, and H. Xiang. Complex spin hamiltonian represented by an artificial neural network, Phys. Rev. B 105, 174422 (2022).
DOI: 10.1103/PhysRevB.105.174422[9] Long range descriptor and potential: H. Yu, L. Hong, S. Chen, X. Gong, and H. Xiang. Capturing Long-Range Interaction with Reciprocal Space Neural Network, arXiv:2211.16684.
arXiv:2211.16684[10] Dielectric response potential: H. Yu, S. Deng, M. Xie, Y. Zhang, X. Shi, J. Zhong, C. He, and H. Xiang. Switchable Ferroelectricity in Subnano Silicon Thin Films, arXiv:2407.01914.
arXiv:2407.01914[11] D3REAM: Guanjian Cheng, Xin-Gao Gong, Wan-Jian Yin. An approach for full space inverse materials design by combining universal machine learning potential, universal property model, and optimization algorithm, Science Bulletin, 2024; 69(19): 3066–3074.
[12] N2AMD: Zhang, C., Zhong, Y., Tao, Z. G., Qin, X., Shang, H., Lan, Z., Prezhdo, O. V., Gong, X. G., Chu, W., Xiang, H. Advancing nonadiabatic molecular dynamics simulations in solids with E(3) equivariant deep neural hamiltonians, Nature Communications 16, 2033 (2025).
[13] Uni-HamGNN-SOC: Zhong, Y., Wang, R., Gong, X., Xiang, H. A Universal Spin-Orbit-Coupled Hamiltonian Model for Accelerated Quantum Material Discovery.
arXiv:2504.19586 (2025)Source Code Repository
Research Areas
Functional Materials Design and Discovery
Leveraging artificial intelligence techniques for efficient prediction and design of novel functional materials, including superconductors, semiconductors, and magnetic materials.
Electronic Structure Prediction
Employing machine learning models to accurately predict materials’ band structures, electron densities, and electronic states, accelerating the exploration of material properties.
Electron-Phonon Coupling Studies
Developing machine learning algorithms to investigate electron-phonon interactions in materials, providing theoretical foundations for superconducting material design.
Magnetic Materials Simulation
Utilizing time-reversal symmetry neural networks to accurately simulate and predict the behaviors and properties of complex magnetic materials.
Latest News
New Paper Published in Nature Computational Science
April 2024
Our research team recently published a paper in Nature Computational Science on a machine learning method to accelerate electron-phonon coupling strength calculations.
Learn MorePlatform Registration Now Open
September 2024
The Laboratory of AI Physical Sciences platform is now open for registration. We welcome researchers worldwide to use our tools to accelerate materials science research.
Register Now